Introduction
The development of a completely new technology does of course not come by itself. Many years on research on a specific molecule, Brivaracetam, has led to the creation of the understanding how to generate crystalline material while flowing heated solution through narrow capillaries. A large part of this research journey has been documented and published in Crystal Growth & Design, also a patent application WO2019215022A1 was submitted based on these results.
In general, our findings pointed out that solutes in solution can start to nucleate very rapidly inside reactors with reduced internal volume. More specifically, as Brivaracetam is a rather complex molecule to crystallize, we observed the possibility of selection a certain polymorphic form of Brivaracetam to nucleate preferentially. The benefits of these long, elongated reactors with narrow inner diameters and even smaller wall thickness are that the solutions are cooled down in a very controlled way from soluble conditions, through the metastable zone, and into the zone where spontaneous nucleation – whether homo- or heterogeneous – may take place. This cooling down is not affected by the fact that we use flow velocities inside these reactors from 10 to 100 cm/s; hence, the use of relevant flow rates – to obtain desired production rates of APIs – becomes possible. Together with the defined cooling of liquids flowing through our reactors, we have also observed to beneficial influence of occurring shear rate inside our reactors on the width of the metastable zone, resulting in a drastic increase in nucleation rates at identical conditions when increasing the shear rates inside the reactors. A side-effect was the observation of the absence of fouling during long-term productions of Brivaracetam from the start. On a side note, we have not considered fouling during the first years of research as we never encountered any blockages of the tubing, even during long runs. In the paragraph discussing fouling and blockages we will go deeper into the reasons behind the fact that blockages inside our reactors will only occur when several critical factors are or are not considered.
Developing a way to crystallize directly the correct crystal size for a solute in solution
From the start, our philosophy was based on the control of the number of nuclei appearing inside a crystallizing slurry. We had observed in literature several attempts to control crystal growth using difficult crystal growth models in different axes of a crystal, the use of CSTR tanks, etc. None of them we found satisfactory for this goal as it complexifies the process and you always rely on the use of solid seeds of some form and shape that results in a redundant process step if crystals are generated spontaneously. On the other hand, if one would be able to control how much nuclei are generated homogeneously while passing through a reactor, then in a simple, maybe even naïve, calculation, one would be able to predict their size once they have grown to their equilibrium state.
Suppose that you would have an initial solution concentration of 400 mg of solute per mL of solvent. And that at a low temperature solubility is 100 mg of solute per mL of solvent. That would mean that per mL of solvent you can crystallize – when performing for this example a cooling crystallization – 300 mg of crystals. Then, if you have 100 crystals in this slurry, or hundred thousand of crystals, this would then result immediately in completely different particle sizes once these crystals are fully grown.
|
Table 1: estimation of particle size of crystals as a function of the total amount of crystals present in a slurry for a given concentration and crystal density.
|
|||
|---|---|---|---|
|
Mass to crystallize
|
300 mg
|
||
|
1 g/mL
|
|||
|
|
|||
|
|
|||
|
Number of crystals
|
volume / mm3
|
Spherical
average length / µm
|
Cubic
average length / µm
|
|
100
|
3
|
895
|
1442
|
|
1000
|
0,3
|
415
|
669
|
|
10000
|
0,03
|
193
|
311
|
|
100000
|
0,003
|
89
|
144
|
|
1000000
|
0,0003
|
42
|
67
|
|
10000000
|
0,00003
|
19
|
31
|
So, for an oral dosage form with average sizes preferentially below the 100 µm mark, anywhere between 105 and 106 crystals produced per mL solvent is to be achieved. Please note that in the above table we assumed both spherical and cubic particles leading to different expected crystal sizes. If we look at estimated nucleation rates for homogeneous crystallization at different conditions we will describe here below, in theory (Mersmann, Crystallization Technology Handbook, 2nd edition, ISBN 0-8247-05828-9, chapter nucleation) a nucleation rate can be expected in between 1012 and 1020 per cubic meter per second, which equals to 106 and 1014 of crystals produced per mL. Comparing both values indicates that whenever we can reach nucleation conditions as set out in Figure 1 and control their evacuation out of the reactors to let the freshly generated nuclei grow in non-nucleating conditions, we would be able to separate nucleation and growth of crystals and target exact reproducible size whenever the passage through our reactors is as homogeneous as possible.
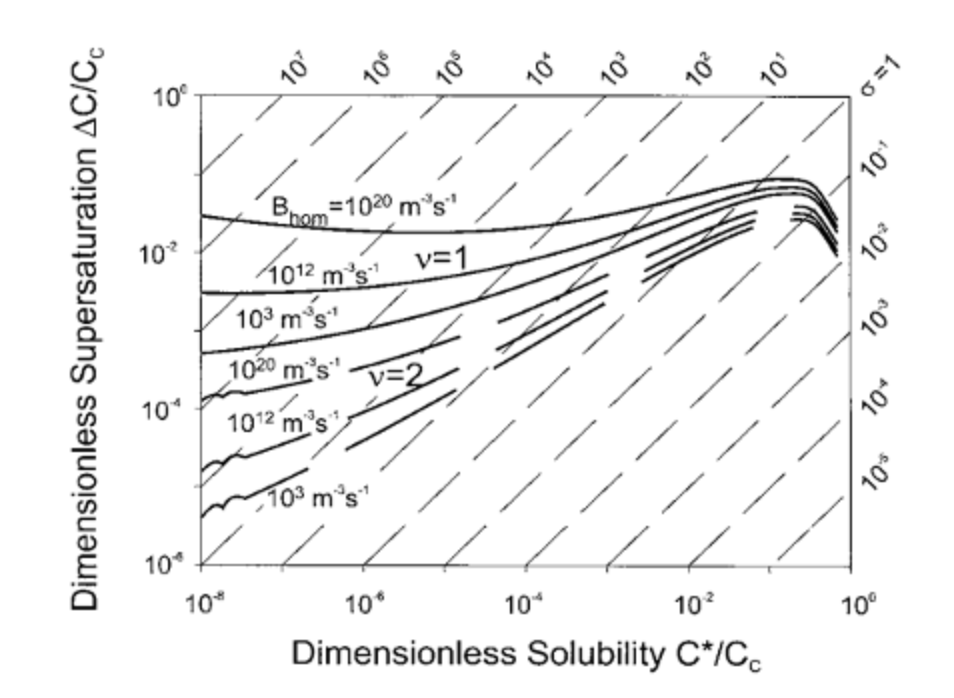
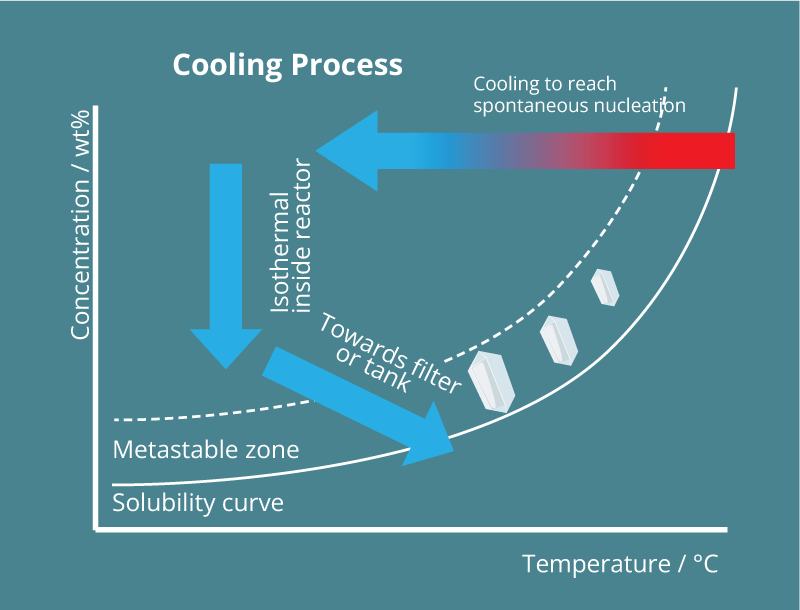
As stated in above figure, solubility and supersaturation are important parameters that rule the occurrence of nucleation. In fact, for a crystalline structure to appear, a treshold energy barrier needs to be overcome in order for the structured material to appear instead of molecules remaining disordered inside the solvent. The nucleation theory is of course already well described, but in general, for a molecule to start nucleating, this energy barrier is most easily depicted using the phase diagram of the solute in solution, as shown in Figure 2. This means that for a solute in solution, to the right of the solid line in Figure 2, needs to be cooled down in order to nucleate. The energy barrier is the most wide for primary homogeneous nucleation, which is nucleation from the solution without any other foreign material present. In the presence of foreign material – impurities, surfaces, etc. – but without any solute-own crystals, primary heterogeneous nucleation takes, with a lower energy barrier to be overcome as for homogeneous nucleation. Once crystals are present in the slurry, these may brake up due to fractures, shear effect, collisions, etc. These are grouped together as secondary nucleation – so with the presence of solute-own crystals in the slurry. Logically, these come at the lowest energy barrier to be overcome.
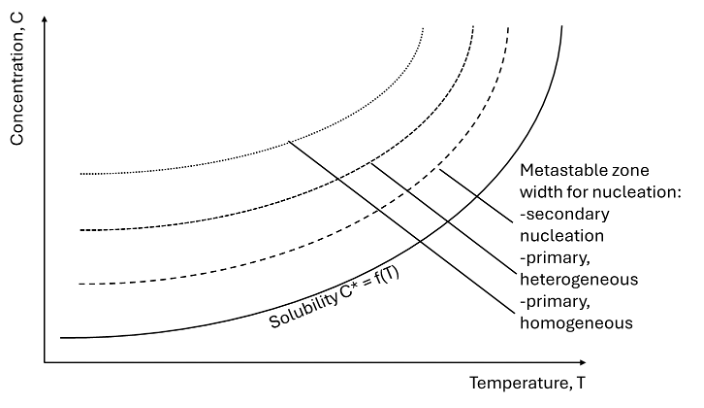
Following our philosophy to control the formation of crystals and to maintain the quantity of crystals nucleated, inside our reactors we aim for primary nucleation, whereas the gathering of the slurry coming out of the reactor has to be done in such a way that secondary nucleation is avoided.
To express this metastable zonewidth, at isothermal conditions supersaturation s is defined as the difference between the concentration C of a system and the solubility concentration at that temperature C*. To create a dimensionless number, this difference between C and C*, is divided by C*:

Once the metastable zonewidth is crossed for primary nucleation, the kinetics of nucleation – the number of nuclei generated – is increased by further cooling, generating a higher supersaturation value. As an example, in Figure 3 the difference in metastable zonewidth is shown between the compounds Brivaracetam and Aspirin in their proper solvents2.
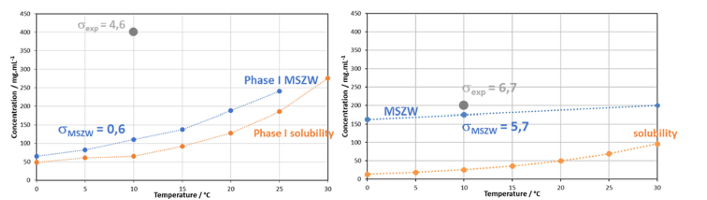
[1] Rimez at al. J Flow Chem 2020, 10, 241-249 (doi: 10.1007/s41981-019-00069-2)
The supersaturation value needed to obtain primary nucleation for Brivaracetam is of 0.6, and experiments can be conducted at concentrations as high as 400 mg/mL, resulting in very high supersaturation values and a large variability in crystal sizes while playing with different conditions can be obtained as published earlier (Rimez et al., Crystal Growth & design 2018, 18, 6431-6439 (doi: 10.1021/acs.cgd.8b00928)). For Aspirin, the metastable zonewidth when dissolved in water/ethanol is much larger, a supersaturation value of 5.7 needs to be overcome to observe nucleation in our reactors, making it for this example difficult to generate working conditions at supersaturations above the optimal value of Brivaracetam (σ = 6.7 instead of 4.6). However, both obtained supersaturations are high and it can indeed be expected that predicted nucleation rates and hence refined particles sizes can be obtained for both examples, with reported particle sizes between 50 and 200 µm for Brivaracetam3 and in used solvent between 400 and 800 µm for Aspirin2. Hence, the key factor to crystallize directly the desired particle size is to nucleate a certain amount of nuclei inside the solution and gather this liquid to let the crystals grow to their equilibrium state, independent of the time required to do so, as long as primary nucleation event only take place inside the specific nucleation reactor. As a side-note: all solution passing through such a reactor needs to undergo the exact same cooling pattern as to homogenize maximally the nucleation rate for the complete production liquid envisaged.
The use of capillary reactors to control the nucleation rate while maintaining uniformity
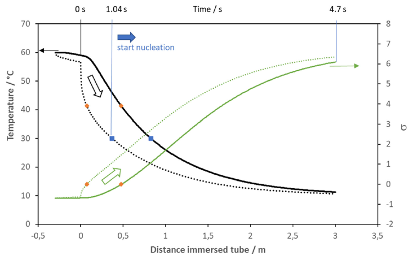
In order to cool a liquid down not only very rapidly but also in a uniform way, we opted for millifluidic tubing with maximally 1 mm inner diameter. We conclude this after an experimental study on Aspirin (Rimez et az., J Flow Chem 2019, 9, 237-249 (doi : 10.1007/s41981-019-00042-z)) where we experimentally and by simulation demonstrated how a liquid cools down when it is injected at a constant rate inside a tubular reactor of 1 mm having a wall thickness of 150 µm and being placed in a water bath of at least 100 times the volume as opposed to the volume of the reactor. For the study on Aspirin, we used a reactor of 3 m length which corresponds roughly to 3 mL of volume; this reactor was placed inside a 1 L water bath that was set at the desired temperature. Then we measured using thermocouples at different distances the temperature inside the reactor while liquid was flowing through the reactor. This liquid was preheated at the desired solution temperature for the Aspirin solution in ethanol/water (50/50 v/v). this overlapped with the simulate values for the tube center temperature. In the next figure, we show the simulated values for both the tube center and the simulated temperature at the tube wall.
In Figure 4 the liquid entering the reactor is indicated at distance zero. The temperature of the liquid drops drastically at the tube wall (dashed black lines), followed by the temperature in the center (solid black line). The dots in orange indicate when the liquid passes the solubility temperature of the concentration of 200 mg/mL; the blue dots correspond to the metastable zonewidth. It can therefore be observed that after a distance shorter than 50 cm inside the tubing the solution is already in conditions where spontaneous nucleation will take place, the remainder of the time inside the tubing, 4.7 s in total at ruling flow velocities, renders the possibility to produce a large quantity of nuclei in order to reach the desired amount of crystals. These short residence times prevent crystals to grow to sizes that might become difficult to handle.
Any crystal growth is therefore also conducted outside of our reactors. In general, the next schematics represent very well how we perform crystallizations, see Figure 5: a heated liquid is injected at a constant rate inside a tubular reactor sitting at a low temperature at which the nucleation should take place. Before this cooling sown there is a possibility to alter flow hydrodynamics inside the tubing or add an antisolvent – these topics will be discussed later. While flowing through the tubing all liquid is always cooled down in exactly the same way: laminar flow conditions are reached for the combination of flow rates and tubing diameter we apply, therefore plug flow conditions are reached and all material is constantly moved forward through the tubing. Inside the phase diagram we go as fast as possible to the desired nucleation temperature (horizontal arrow from high to low temperatures in the phase diagram). At this point, the nucleation is already started, indicating a drop in solute concentration (vertical arrow in the phase diagram) while the liquid is still flowing throught the tubing. Then, at the end of the tubing, the slurry containing fast growing crystals can be gathered inside a waiting tank for the crystal growth to be fulfilled or, when the growth kinetics allow this, directly on a filtration unit. For crystal growth conditions, any temperature might be selected.
Benefits of capillaries: solid particle mobility and shear rates
The uniformity in the cooling of liquids flowing through the tubing but also the development of a laminar flow regime inside the capillaries is beneficial for SCT. As the estimated Reynolds numbers are well below 4000, see Table 2, generally considered the threshold for turbulent flow, laminar flow behavior is to be expected. On the other hand, capillary reactors possess other beneficial aspects that are in use inside our reactors.
|
Table 2: typical flow rates with corresponding flow velocity inside capillary reactors and estimated Reynolds number using water at room temperature as basis for calculation.
|
||||
|---|---|---|---|---|
|
Flow rate
|
mL/min
|
10
|
20
|
30
|
|
Flow velocity
|
cm/s
|
21,2
|
42,4
|
63,7
|
|
Reynolds number
|
|
20
|
400
|
600
|
|
Shear rate
|
s-1
|
1698
|
3395
|
5093
|
At usual flow rates inside the reactors, shear rates at the tube are above 1000 s-1. For other types of solid particles flowing through narrow channels under laminar flow conditions and elevated shear rates, it is known that these particles do not migrate towards the channel wall. Most famously is the Fåhræus–Lindqvist effect that describes the migration of erythrocytes to the center of the vessel, leaving only plasma neat the wall of the vessel (https://en.wikipedia.org/wiki/F%C3%A5hr%C3%A6us%E2%80%93Lindqvist_effect). In Figure 6 an artificial vein is shown with hemoglobin flowing through, showcasing the absence of hemoglobine at the tube wall.

When simulating the preferential position of solid particles inside circular tubings under constant flow conditions, Javier Rivero-Rodriguez and Prof. Benoit Scheid of the Université libre de Bruxelles (Rivero-Rodriguez and Scheid, J Fluid Mech 2018, 842, 215-247 (doi: 10.1017/jfm.2018.78)) discovered that small particles, when not subjected to gravity, will migrate under laminar flow conditions to a preferentially eccentric position, away from the tube center. At the tube wall the lowest probability to retrieve a solid particle is observed. This preferential position is shown in Figure 7. It is demonstrated that the difference in flow velocity in the laminar flow regime pushes the particle away from the tube center. On the other hand, the particles bounce off the tube wall, generating an equilibrium position off center. Therefore, as long as the particles are not subjected to gravity, the flow conditions inside of the reactors prevent collisions and deposition on the tube wall, resulting on blockages of the tubing.
Using Secoya’s Crystallization Technology, a large amount of nuclei are however formed at different conditions, which may provoke coagulation of these particles, very fast crystal growth, etc. Therefore, the use of a certain length for the reactor depends on the trade-off between sufficiently high nucleation rates and avoiding the possibility to block the tubing during operation due to too intense growth for example. The study to find the optimal reactor dimensions is an integral part of the development cycle of the process at the laboratory scale.
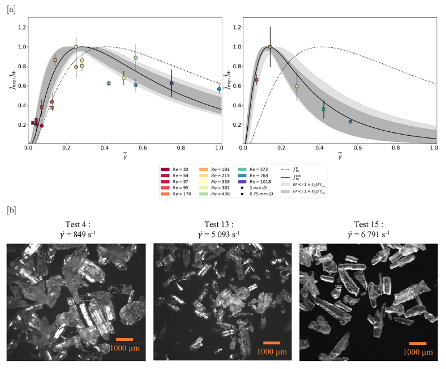
Another benefit of the tubular reactors is the high shear rates obtained inside. Mura and Zaccone have described mathematically the preferential nucleation conditions for particles under flow conditions and high shear rates (Mura and Zaccone, Phys Rev E 2016, 93, 042803 (doi: 10.1103/physRevE.93.042803)). in a joint study between Zaccone, ULB and Secoya, experimental data further evidenced this beneficial effect of shear rate in flow on the nucleation rate of a solute in solution (Debuysschère et al., Cryst Growth Des. 2023, 23, 4979-4989). In this work a cooling crystallization of Glycine in water was conducted. All Thermodynamic variables were kept constant (solution concentration and temperature, residence time at low temperature, nucleation temperature, etc.). The only variable was a change in shear rate while flowing through the reactor. This was achieved by varying the capillary diameter, flow rate and reactor lengths to obtain always the same thermodynamic conditions. The result was that the measured nucleation rate, shown in Figure 8a goes through an optimal value at a shear rate of 3000 s-1 and Reynolds number of 340 for 98% pure Glycine, whereas 99.5+ % pure Glycine had its maximum at a slightly lower shear rate of 1700 s-1. Other evidences on the influence of more localized shear rates generated inside the tubing in different studies (different studies already referenced here above) lead to further evidencing the large impact of shear rates on the nucleation rate of a solute in solution while cooling down. For any cooling crystallization study, the shear rate influence is part of the development cycle for optimization of the crystallization of any molecule.
Conclusions
Secoya’s Crystallization Technology is based on the adaptation of the size and shape of tubular reactors to the speed of nucleation of solutes in solution. The overall short residence times and uniform cooling conditions inside the reactors result in the control of primary nucleation, enabling a prediction of the final particle size of crystalline material after complete growth of the crystals, outside the reactor in a collecting vessel. Focusing only on nucleation in flow conditions also avoids blockages during production, as nuclei inside the reactors will not be able to migrate to the tube wall resulting in blocked tubings. The high shear rates obtained inside the reactors have an extra beneficial effect on the nucleation rate.


